s********9
发帖数: 132 | 1 文章里面似乎没有怎么说. 我自己不是搞结构的,只是想学习一点.
尤其是surface的展示比较好的.
谢谢. |
f**********1
发帖数: 203 | 2 Pymol
VMD
molmol
【在 s********9 的大作中提到】
: 文章里面似乎没有怎么说. 我自己不是搞结构的,只是想学习一点.
: 尤其是surface的展示比较好的.
: 谢谢.
|
s********9
发帖数: 132 | 3 我稍微学了些jmol和SPDV.你说的这些是不是比这两个要强大(所以专业的结构生物学家写文章用这
些)? 你觉得哪个界面比较友好,适合我这种业余选手?
谢谢
【在 f**********1 的大作中提到】
: Pymol
: VMD
: molmol
|
s********9
发帖数: 132 | 4 附件里面的一个图是spdv官网的turial里面的例子,我试了半天也没有能搞成那样.
另一个图片是从发表的文章里面截的图,对几个软件都熟悉的人能看出是什么软件搞的
么?
谢谢
家写文章用这
【在 s********9 的大作中提到】
: 我稍微学了些jmol和SPDV.你说的这些是不是比这两个要强大(所以专业的结构生物学家写文章用这
: 些)? 你觉得哪个界面比较友好,适合我这种业余选手?
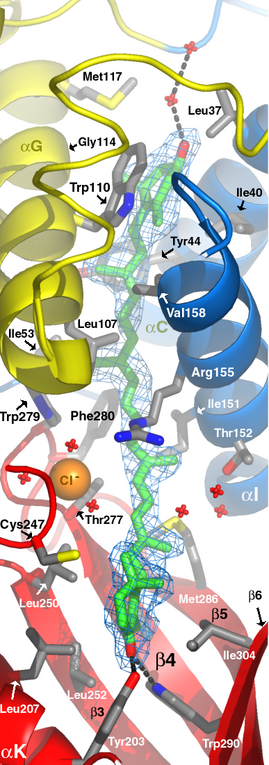
: 谢谢
|
s*****g
发帖数: 87 | |
f**********1
发帖数: 203 | 6 Snap1.jpg是Pymol,
绿色是它们默认的颜色
【在 s********9 的大作中提到】
: 附件里面的一个图是spdv官网的turial里面的例子,我试了半天也没有能搞成那样.
: 另一个图片是从发表的文章里面截的图,对几个软件都熟悉的人能看出是什么软件搞的
: 么?
: 谢谢
:
: 家写文章用这
|
f**********1
发帖数: 203 | 7 我个人比较喜欢pymol,界面友好,简单好用,还可以用Python scripting,
比较容易上手
VMD功能比较强大,option很多,初用者可能得花一些时间才能熟悉
家写文章用这
【在 s********9 的大作中提到】
: 我稍微学了些jmol和SPDV.你说的这些是不是比这两个要强大(所以专业的结构生物学家写文章用这
: 些)? 你觉得哪个界面比较友好,适合我这种业余选手?
: 谢谢
|
p*****u
发帖数: 191 | 8 VMD也可以Python
【在 f**********1 的大作中提到】
: 我个人比较喜欢pymol,界面友好,简单好用,还可以用Python scripting,
: 比较容易上手
: VMD功能比较强大,option很多,初用者可能得花一些时间才能熟悉
:
: 家写文章用这
|
|
s********9
发帖数: 132 | 9 综合几位高手的发言,我就打算先搞vmd了.
spdv的turial的作者说spdv是最popular的软件,结果看来不是那么回事阿.我一个笨人学点
东西容易么我.
谢谢.
【在 p*****u 的大作中提到】
: VMD也可以Python
|
l**********1
发帖数: 5204 | 10 Sure
先下载个VMD 试一下吧
link:
//www.ks.uiuc.edu/Research/vmd/plugins/pythonlib/
also one fancy Figure
captured from
//www.ks.uiuc.edu/
人学点
【在 s********9 的大作中提到】
: 综合几位高手的发言,我就打算先搞vmd了.
: spdv的turial的作者说spdv是最popular的软件,结果看来不是那么回事阿.我一个笨人学点
: 东西容易么我.
: 谢谢.
|
|
|
V**3
发帖数: 12756 | 11 这图Pymol做的很轻松啊
有没有大侠介绍一下vmd 比pymol 优势的地方,比如哪些功能?
一定包子酬谢
【在 l**********1 的大作中提到】
: Sure
: 先下载个VMD 试一下吧
: link:
: //www.ks.uiuc.edu/Research/vmd/plugins/pythonlib/
: also one fancy Figure
: captured from
: //www.ks.uiuc.edu/
:
: 人学点
|
p*****u
发帖数: 191 | 12 我个人觉得,其实功能相差无多大异处。VMD的诞生是为了动力学模拟;Pymol的诞生当
初是为了view分子。现在的功能大部分集中到了插件上了。如今双方的实力都很牛,风
格也迥然不同;VMD和Pymol的扩展性都很好,想要更好更多的功能就看谁拥有的插件多
。另外,VMD能够利用CUDA加速阿。
分享一个由VMD做的太极拳表演,娱乐娱乐:
//v.youku.com/v_show/id_XMTU0NjA5MjU2.html
【在 V**3 的大作中提到】
: 这图Pymol做的很轻松啊
: 有没有大侠介绍一下vmd 比pymol 优势的地方,比如哪些功能?
: 一定包子酬谢
|
C*******I
发帖数: 151 | |
p*****u
发帖数: 191 | 14 你是说20年前吧?
看看它们的gallery,然后跟BallView, ICM-Browser, Chimera比较一下
【在 C*******I 的大作中提到】
: 图片效果最好的应该是Molscript/Raster3D
: Molscrip:http://www.avatar.se/molscript/
: Raster3D:http://skuld.bmsc.washington.edu/raster3d/
: 用法:
: http://people.mbi.ucla.edu/sawaya/tutorials/Graphics/render.htm
|
s********9
发帖数: 132 | 15 而且VMD是免费的.pymol现在收费了.
再问下:
1:homology model用哪个好? SPDV可以,不知道哪个好些? 文献上面说的homology的定量(比
如40% 60%是怎么来的? 是根据structural seq alignment 数完全相同的氨基酸数量除以总
数?还是别的?clustalw2是不给量化结果的.)
2:1)关于氢键,做结构的人文章里面到底donor atom 和acceptor atom离多远才算是可信? 我
前面用spdv似乎是定在3.5A.可是看一个文章里面描述的一个结构里面所谓重要的氢键已经4A以上
了.
2)一般说是N(O)-H...N(O),可是又看到有的文章里面说说C-H...N(o),这个氢键难道就是个
任人打扮的小姑娘?
3)如果在结构里面看到某个氨基酸的side chain和别的氨基酸有H bond,而我打算mutate这个
amino acids,一个结构生物学的人会怎么去推测这个mutation whether or to what
extent will affect the overall structure of the motif.
非常感谢各位!
【在 s********9 的大作中提到】
: 文章里面似乎没有怎么说. 我自己不是搞结构的,只是想学习一点.
: 尤其是surface的展示比较好的.
: 谢谢.
|
C*******I
发帖数: 151 | 16 历史久不是缺点吧?molscript/raster3D的网站的图集根本没代表性。这个package色彩质感各项
参数自由度非常大,这个要自己去调整体会。用它发表文章的图片大多都比那些图集好得多。只有下面这
一张还有点体现molscript/raster3D 的功能。
(http://people.mbi.ucla.edu/sawaya/tutorials/Graphics/pymol2.png)
现在有些软件也不错。chimera历史很久也了,功能也比过去强很多了。我有时候也用,但图片效果还
是不如molscript/raster3D。用molscript/raster3D作movie不方便的时候 我也用
chimera。
【在 p*****u 的大作中提到】
: 你是说20年前吧?
: 看看它们的gallery,然后跟BallView, ICM-Browser, Chimera比较一下
|
f**********1
发帖数: 203 | 17 这个图片是pymol做的吧
色彩质感各项
好得
用,但图片效果还
【在 C*******I 的大作中提到】
: 历史久不是缺点吧?molscript/raster3D的网站的图集根本没代表性。这个package色彩质感各项
: 参数自由度非常大,这个要自己去调整体会。用它发表文章的图片大多都比那些图集好得多。只有下面这
: 一张还有点体现molscript/raster3D 的功能。
: (http://people.mbi.ucla.edu/sawaya/tutorials/Graphics/pymol2.png)
: 现在有些软件也不错。chimera历史很久也了,功能也比过去强很多了。我有时候也用,但图片效果还
: 是不如molscript/raster3D。用molscript/raster3D作movie不方便的时候 我也用
: chimera。
|
C*******I
发帖数: 151 | 18 现在pymol也做得出来这样的smog效果了?那我要回去再看看了。
【在 f**********1 的大作中提到】
: 这个图片是pymol做的吧
:
: 色彩质感各项
: 好得
: 用,但图片效果还
|
x*****g
发帖数: 42 | 19 pymol还是开源的吧? 以前也收费的.
定量(比
除以总
可信? 我
键已经4A以上
道就是个
【在 s********9 的大作中提到】
: 而且VMD是免费的.pymol现在收费了.
: 再问下:
: 1:homology model用哪个好? SPDV可以,不知道哪个好些? 文献上面说的homology的定量(比
: 如40% 60%是怎么来的? 是根据structural seq alignment 数完全相同的氨基酸数量除以总
: 数?还是别的?clustalw2是不给量化结果的.)
: 2:1)关于氢键,做结构的人文章里面到底donor atom 和acceptor atom离多远才算是可信? 我
: 前面用spdv似乎是定在3.5A.可是看一个文章里面描述的一个结构里面所谓重要的氢键已经4A以上
: 了.
: 2)一般说是N(O)-H...N(O),可是又看到有的文章里面说说C-H...N(o),这个氢键难道就是个
: 任人打扮的小姑娘?
|
s********n
发帖数: 2939 | |
|
|
p*****u
发帖数: 191 | 21 时代是在进步的,你给的例子确实是pymol做的。
目前的这些软件都很自由,硬件发展快,这些软件也很牛;就拿Chimera来说,你想要
的那种自由设置也有,唯一不方便改变的就是他的风格。但是质量没有任何问题。
Molscript老了,历史已久,并且10多年前就停止了开发和更新,而新的软件不断
出现和更新,说超越,一点都不过分吧。
Raster3D的唯一好处是渲染的时候不依赖于显卡,可是现在的VMD啊, pymol啊,
经过第三方povray的渲染一样不依赖于显卡的。调整好了,质量绝对上乘。
色彩质感各项
好得多。只有下面这
用,但图片效果还
【在 C*******I 的大作中提到】
: 历史久不是缺点吧?molscript/raster3D的网站的图集根本没代表性。这个package色彩质感各项
: 参数自由度非常大,这个要自己去调整体会。用它发表文章的图片大多都比那些图集好得多。只有下面这
: 一张还有点体现molscript/raster3D 的功能。
: (http://people.mbi.ucla.edu/sawaya/tutorials/Graphics/pymol2.png)
: 现在有些软件也不错。chimera历史很久也了,功能也比过去强很多了。我有时候也用,但图片效果还
: 是不如molscript/raster3D。用molscript/raster3D作movie不方便的时候 我也用
: chimera。
|
p*****u
发帖数: 191 | 22 pymol是user surpported的,你依然能下载到源码,并且自己编译。如果自己不想编译
,我觉得你可以去下载bioblender,里面有个pymol 1.3可以提出来用.
说说你的问题
1, 我觉得homology modeling用modeller就很好。而且最近出了一个Pymod将n多软件集
成,建议你用一下;当然前提是clustalw modeller等先预装的。
至于序列相似性程度,假如A含有300个AA,而B是A中一个类似的domain;只含有100
个AA;这个相似性就不用依赖于两个蛋白aa的数量完全相同了吧。另外我感觉,两个蛋
白进行alignment,很多软件得出的结果差不多,只不过使用的算法不一样,pymol也可
以alignment的,看了一下代码;人家用的是交叉熵算法;挺时髦的。
2,氢键的定义吧,我觉得很多文献中都会给出定义的,energy都给出了一定的总结。如
果你用VMD做氢键,就会觉得很痛苦,必须要求结构中有氢原子,这点跟pymol是不同的
,而且vmd的作者也坚持,就根据氢键的定义来:“没有氢怎么会有氢键”。但是也有
人说,一定的距离内,H可以被诱导,形成氢键。当然你说的4.0A就比较玄乎了。
定量(比
除以总
可信? 我
键已经4A以上
道就是个
【在 s********9 的大作中提到】
: 而且VMD是免费的.pymol现在收费了.
: 再问下:
: 1:homology model用哪个好? SPDV可以,不知道哪个好些? 文献上面说的homology的定量(比
: 如40% 60%是怎么来的? 是根据structural seq alignment 数完全相同的氨基酸数量除以总
: 数?还是别的?clustalw2是不给量化结果的.)
: 2:1)关于氢键,做结构的人文章里面到底donor atom 和acceptor atom离多远才算是可信? 我
: 前面用spdv似乎是定在3.5A.可是看一个文章里面描述的一个结构里面所谓重要的氢键已经4A以上
: 了.
: 2)一般说是N(O)-H...N(O),可是又看到有的文章里面说说C-H...N(o),这个氢键难道就是个
: 任人打扮的小姑娘?
|
p*****u
发帖数: 191 | 23 一般说单个残基的对整个蛋白的折叠不会有很大改变,如果不放心,可以做个圆二色谱
来看看。
突变残基的构象选取在pymol中有一个简单的评价,这个如同楼上有人说coot也不错,
coot里面也是有评价的。
定量(比
除以总
可信? 我
键已经4A以上
道就是个
【在 s********9 的大作中提到】
: 而且VMD是免费的.pymol现在收费了.
: 再问下:
: 1:homology model用哪个好? SPDV可以,不知道哪个好些? 文献上面说的homology的定量(比
: 如40% 60%是怎么来的? 是根据structural seq alignment 数完全相同的氨基酸数量除以总
: 数?还是别的?clustalw2是不给量化结果的.)
: 2:1)关于氢键,做结构的人文章里面到底donor atom 和acceptor atom离多远才算是可信? 我
: 前面用spdv似乎是定在3.5A.可是看一个文章里面描述的一个结构里面所谓重要的氢键已经4A以上
: 了.
: 2)一般说是N(O)-H...N(O),可是又看到有的文章里面说说C-H...N(o),这个氢键难道就是个
: 任人打扮的小姑娘?
|
s********9
发帖数: 132 | 24 能具体说一下那个homology打分吗?
比如A和B蛋白有个同源domain50个aa,怎么看计算这个同源性是百分之多少?发表的结构生物学的文章
里面是用什么软件计算的?
100
【在 p*****u 的大作中提到】
: pymol是user surpported的,你依然能下载到源码,并且自己编译。如果自己不想编译
: ,我觉得你可以去下载bioblender,里面有个pymol 1.3可以提出来用.
: 说说你的问题
: 1, 我觉得homology modeling用modeller就很好。而且最近出了一个Pymod将n多软件集
: 成,建议你用一下;当然前提是clustalw modeller等先预装的。
: 至于序列相似性程度,假如A含有300个AA,而B是A中一个类似的domain;只含有100
: 个AA;这个相似性就不用依赖于两个蛋白aa的数量完全相同了吧。另外我感觉,两个蛋
: 白进行alignment,很多软件得出的结果差不多,只不过使用的算法不一样,pymol也可
: 以alignment的,看了一下代码;人家用的是交叉熵算法;挺时髦的。
: 2,氢键的定义吧,我觉得很多文献中都会给出定义的,energy都给出了一定的总结。如
|
s********9
发帖数: 132 | 25 我们不做圆二色谱,没有这个东西,也不会做.就是基于现有的vmd这些软件是否可以给些
speculation.担心是因为像帖子里面说的已经看到这个侧链和别的氨基酸之间有氢键作
用.而且做点突变大多数时候都是极性aa,出现这种情况可能性还是挺大的.
非常感谢
【在 p*****u 的大作中提到】
: 一般说单个残基的对整个蛋白的折叠不会有很大改变,如果不放心,可以做个圆二色谱
: 来看看。
: 突变残基的构象选取在pymol中有一个简单的评价,这个如同楼上有人说coot也不错,
: coot里面也是有评价的。
:
: 定量(比
: 除以总
: 可信? 我
: 键已经4A以上
: 道就是个
|
p*****u
发帖数: 191 | 26 Sequences identities 和 similarities这个不用说吧
不要把打分看得那么重要
构生物学的文章
【在 s********9 的大作中提到】
: 能具体说一下那个homology打分吗?
: 比如A和B蛋白有个同源domain50个aa,怎么看计算这个同源性是百分之多少?发表的结构生物学的文章
: 里面是用什么软件计算的?
:
: 100
|
p*****u
发帖数: 191 | 27 如果有兴趣,可以看看这个综述
Visualization of macromolecular structures
Nature Methods 7,S42–S55(1 March 2010) | doi:10.1038/nmeth.1427
//www.nature.com/nmeth/journal/v7/n3s/full/nmeth.1427.html
【在 C*******I 的大作中提到】
: 现在pymol也做得出来这样的smog效果了?那我要回去再看看了。
|
C*******e
发帖数: 4348 | 28 最新一版的pymol图片效果很不错的
今年1月才发布的
【在 C*******I 的大作中提到】
: 现在pymol也做得出来这样的smog效果了?那我要回去再看看了。
|
C*******e
发帖数: 4348 | 29 现在分licence版和免费版
【在 x*****g 的大作中提到】
: pymol还是开源的吧? 以前也收费的.
:
: 定量(比
: 除以总
: 可信? 我
: 键已经4A以上
: 道就是个
|
C*******e
发帖数: 4348 | |
|
|
s********9
发帖数: 132 | 31 这次附了两个图:
1)snap3 shows the N-C-H...O hbond longer than 4A, which is claimed be to
critical in that structure.
2)snap4 shows that mutation I was talking about. See that hydrogen bond
(3.
13A) is between two sheets. If we mutate that to non-polar aa, can you
give
any speculations?
3)SNAP5 是我自己用vmd represent的electrostatic potential surf 和peptide
binding的图(非常感谢前面回帖的所有人,我总算蜗牛般地学了点vmd).
snap6是文献里面的同样的展示内容.
困扰我的是我做的里面那个binding site像个闭合的孔,而文献里面四个半开放的
channel.
我的surf不是因为计算的electrostaic potential的问题,因为如图SNAP7,我即使只做
surf,而不要potential,那个binding site 还是闭合的.能不能指点一下问题所在?
4)追问一下:homology modelling,在
modeller,jmol,spdv,vmd这几个里面,哪个更被专业的人认可(活着jmol和vmd根本没有这个功
能?)?
非常非常感谢
【在 p*****u 的大作中提到】
: Sequences identities 和 similarities这个不用说吧
: 不要把打分看得那么重要
:
: 构生物学的文章
|
s********9
发帖数: 132 | 32
give
【在 s********9 的大作中提到】
: 这次附了两个图:
: 1)snap3 shows the N-C-H...O hbond longer than 4A, which is claimed be to
: critical in that structure.
: 2)snap4 shows that mutation I was talking about. See that hydrogen bond
: (3.
: 13A) is between two sheets. If we mutate that to non-polar aa, can you
: give
: any speculations?
: 3)SNAP5 是我自己用vmd represent的electrostatic potential surf 和peptide
: binding的图(非常感谢前面回帖的所有人,我总算蜗牛般地学了点vmd).
|
s********9
发帖数: 132 | 33
【在 s********9 的大作中提到】
: 这次附了两个图:
: 1)snap3 shows the N-C-H...O hbond longer than 4A, which is claimed be to
: critical in that structure.
: 2)snap4 shows that mutation I was talking about. See that hydrogen bond
: (3.
: 13A) is between two sheets. If we mutate that to non-polar aa, can you
: give
: any speculations?
: 3)SNAP5 是我自己用vmd represent的electrostatic potential surf 和peptide
: binding的图(非常感谢前面回帖的所有人,我总算蜗牛般地学了点vmd).
|
p*****u
发帖数: 191 | 34 感觉文献的比你的缺了点东西。
你不会为这么小的肽段做homology modeling吧
试着修改一下probe的大小 |
{kind=link}